Protein phosphorylation controls nearly every biological process and is dsyregulated in a host of human diseases, including immune disorders, neurodegenerative disorders, and cancers. Phosphorylation is performed by a class of protein enzymes called kinases. In humans, there are ~300 serine/threonine kinases that collectively phosphorylate more than 90,000 sites distributed across virtually every protein. Our knowledge of this extraordinary complexity has so far been limited – only a small number of these phosphorylation sites (<4%) has been linked to its kinase. Determining how all this information is organized into cellular signaling networks is necessary for understanding biological processes and diseases.
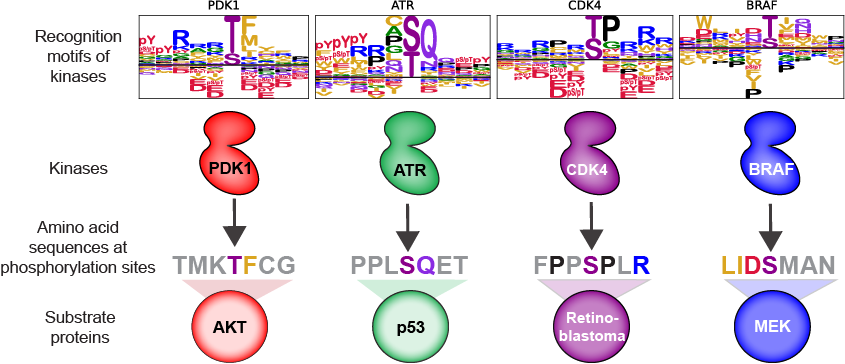
The Cantley lab has shown that the logic behind these vast signaling networks can be distilled down to simple rules. Protein kinases are guided by ‘codes’ in deciding which of the thousands of proteins to target for phosphorylation. They are tuned to recognize specific sequences of amino acids surrounding their precise target sites. This is often referred to as a kinase’s recognition motif, and it tells us how well a kinase (“key”) will match to a given target site (“lock”) on a protein. In recently published work, Jared Johnson (instructor) and Tomer Yaron (graduate student) in the Cantley Lab, in collaboration with the Yaffe and Turk Laboratories, determined the recognition motifs for nearly every serine/threonine kinase encoded in the human genome. The authors then used this information to accurately predict the most likely protein kinase to phosphorylate any site in a human protein based on its amino acid sequence. Their work reveals the first complete characterization of the substrate specificity of the human serine/threonine kinome and provides a valuable resource (available in the following link as a webtool: https://kinase-library.phosphosite.org/site) for researchers studying signalling in human biology and disease.