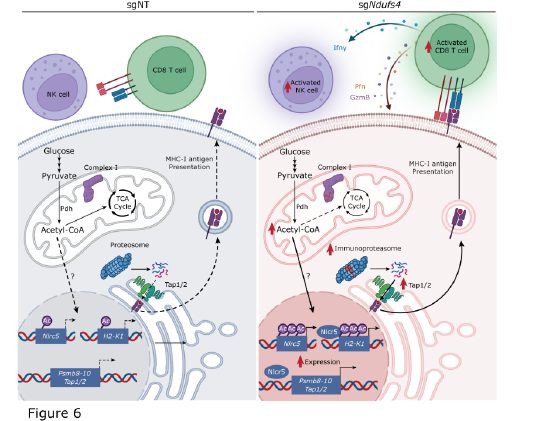
While over the last few years immune checkpoint blockade, therapeutics that take the breaks off of immune cells to fight cancer have revolutionized how we treat the disease, many people still do not meaningfully respond to therapy. Others have found that cancer cells frequently rewire their metabolism to support proliferation and evade immune surveillance, but little is known about metabolic targets that could increase immune surveillance. In this paper, we show that a specific means of mitochondrial respiratory complex I (CI) inhibition improves tumor immunogenicity and sensitivity to immune checkpoint blockade (ICB). Targeted genetic deletion of either Ndufs4 or Ndufs6, but not other CI subunits, induces an immune-dependent growth attenuation in melanoma and breast cancer models. We show that deletion of Ndufs4 induces expression of the major histocompatibility complex (MHC) class I co-activator Nlrc5 and antigen presentation machinery components, most notably H2-K1. This induction of MHC-related genes is driven by a pyruvate dehydrogenase-dependent accumulation of mitochondrial acetyl-CoA, which leads to an increase in histone H3K27 acetylation within the Nlrc5 and H2-K1 promoters. Taken together, this work shows that selective CI inhibition restricts tumor growth and that specific targeting of Ndufs4 or Ndufs6 increases T cell surveillance and ICB responsiveness and opens the door for new mitochondrially targeted therapies.