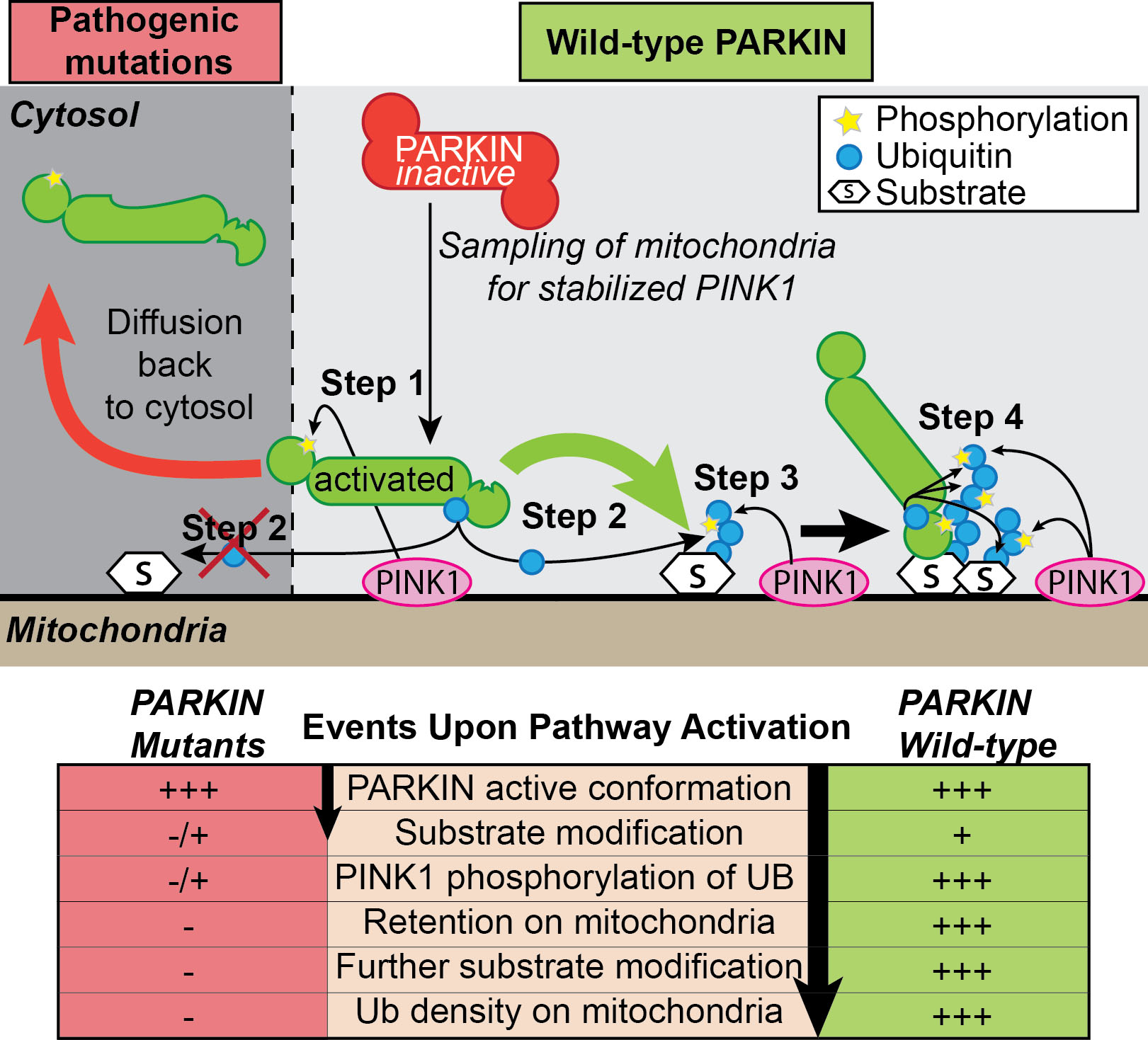
PINK1 and PARKIN – two proteins mutated in early onset Parkinson’s Disease - are known to function in a signaling cascade that leads to ubiquitylation of mitochondrial outer membrane proteins on damaged mitochondria, but the precise mechanism through by which PINK1 activates PARKIN ubiquitin ligase activity and retention on the mitochondrial membrane is poorly understood. In the most recent issue of Molecular Cell,Alban Ordureau in the Harper Lab used quantitative proteomics and a technique called ubiquitin AQUA to examine the kinetics and specificity of PINK1 and PARKIN-dependent ubiquitin chain synthesis on damaged mitochondria in vivo. Through mechanistic and biochemical analysis, as well as live-cell imaging, the authors define multiple steps in the process, revealing a feed-forward mechanism for PARKIN activation and retention on mitochondria. PINK1 phosphorylation of S65 in the UBL of PARKIN leads to activation of its ubiquitin ligase activity by 2400-fold, which in turn promotes the initial synthesis of K6, K11, K48, and K63 ubiquitin chains on mitochondria by PARKIN. Ubiquitin units within newly synthesized ubiquitin chains are then phosphorylated by PINK1 on S65, the residue homologous to S65 in the UBL of PARKIN. This, in turn, serves as a binding site (Kd = 17 nM) for activated PARKIN, leading to retention of PARKIN on poly-ubiquitinated mitochondria, which could support both further ubiquitin chain synthesis and recruitment of proteins to promote mitophagy. Our data reveal a feed-forward mechanism that explains how PINK1 phosphorylation of both PARKIN and poly-UB chains synthesized by PARKIN drives a program of PARKIN recruitment and mitochondrial ubiquitylation in response to mitochondrial damage. This work also provides a framework for quantitative analysis of phosphorylation and ubiquitin dependent signaling systems in vitro and in vivo.