In collaboration with the Shao Lab (HMS Cell Biology) and the Elledge Lab (HMS Genetics), the Gu Lab delves deeper into the molecular mechanism and pathophysiological relevance of midnolin using structural biology, biochemistry, and cell biology approaches.
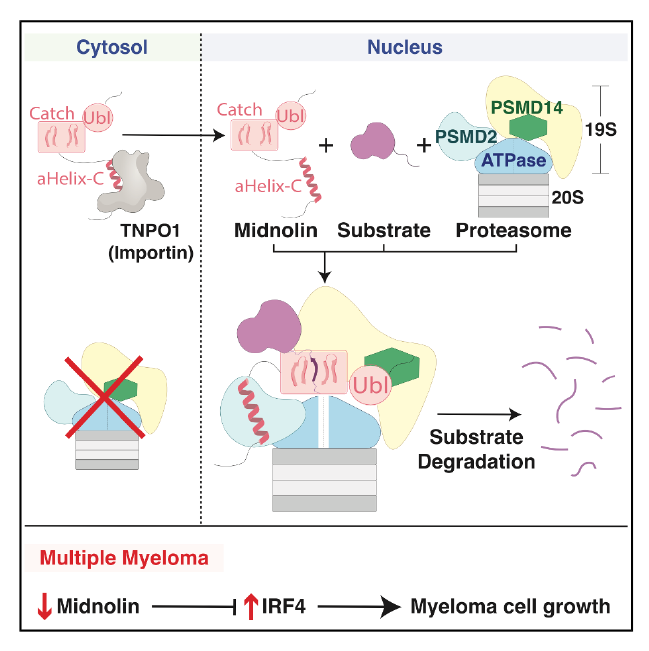
The midnolin-proteasome pathway degrades many nuclear proteins without ubiquitination, but how it operates mechanistically remained unclear. Here, we present structures of the midnolin-proteasome complex, revealing how established proteasomal components are repurposed to enable a unique form of proteolysis. While the proteasomal subunit PSMD2/Rpn1 binds to ubiquitinated or ubiquitin-like (Ubl) proteins, we discover that it also interacts with the midnolin nuclear localization sequence. We show that importins and proteasomes compete for the midnolin nuclear localization sequence, enabling the midnolin-proteasome complex to form only in the nucleus. This result clarifies our previous observations on how midnolin’s activity is confined to the nucleus.
Furthermore, PSMD14/Rpn11, an enzyme that normally cleaves ubiquitin chains, surprisingly functions non-enzymatically as a receptor for the midnolin Ubl domain. This interaction positions the substrate-binding Catch domain directly above the proteasomal entry site to guide substrates into the proteasome.
Finally, we demonstrate midnolin expression is downregulated in multiple myeloma, both in cell lines and in patient-derived plasma cells. This downregulation occurs early in disease progression, before plasma cells transition to malignancy. Remarkably, re-expressing midnolin in myeloma cells halts proliferation through the degradation of a single transcription factor, IRF4.
Overall, our findings uncover the mechanisms underlying the midnolin-proteasome pathway and midnolin downregulation as a driver of multiple myeloma.