About 5-10% of all cancers are linked to inherited mutations in tumor suppressor genes. People born with these mutations face a much higher risk of developing cancers, often at an earlier age. For example, a mutation in the BRCA1 gene can lead to up to an 80% lifetime risk of breast cancer. This increased risk has been conventionally explained by the “two-hit” hypothesis. It reasons that it would take less time to fully inactivate a tumor suppressor gene in individuals born with only one functional allele compared to those with two alleles, thus speeding up cancer development in heterozygous mutation carriers.
However, a new study from the Brugge Lab, led by postdoctoral fellow Carman Li, provides evidence of an additional mechanism driving cancer risk. By creating genetically engineered mice with either a Brca1 mutation (germline heterozygous) or no mutation (wild-type), the team discovered that even when Brca1 was deleted in both groups simultaneously, mice with the germline mutation still developed tumors earlier than their wild-type counterpart, contrary to the prediction of the “two-hit” model. This suggests that the heterozygous state itself primes cells for cancer initiation, independent of the timing of full Brca1 loss.
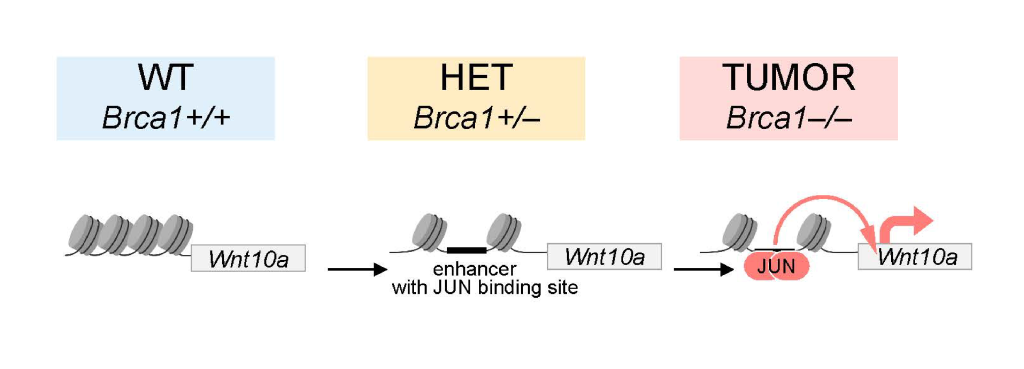
Further analysis using single-cell RNA sequencing and ATAC-seq chromatin profiling revealed unusual epigenetic changes in Brca1-heterozygous mammary cells, with patterns of increased chromatin accessibility that resemble those found in cancer cells. These alterations involve the transcription factor family AP-1 and a cancer-promoting gene Wnt10a. This "primed" state in Brca1-heterozygous cells could make them more likely to progress to cancer, offering a potential new angle for early intervention by targeting this priming effect.